Maladie polykystique rénale autosomique dominante (MPRAD) :
Une revue à l’intention des médecins de soins primaires
 |
Ahsan Alam, M.D., C.M., M. Sc., FRCPC Professeur agrégé de médecine Division de néphrologie et Programme de multitransplantation Hôpital Royal Victoria – Centre universitaire de santé McGill Montréal (Québec) |
La maladie polykystique rénale autosomique dominante (MPRAD) est la plus fréquente cause héréditaire d’insuffisance rénale; elle occupe le quatrième rang des raisons pour lesquelles les patients en viennent à recourir à la dialyse. La prévalence estimée de la MPRAD se situe entre 1 à 400 et 1 à 1 000 naissances vivantes, ce qui porterait à plus de 30 000 le nombre de Canadiennes et de Canadiens atteints. Selon cette estimation, la MPRAD serait plus répandue que le syndrome de Down, l’hémophilie A, la maladie de Huntington et la fibrose kystique combinés4.
On connaît actuellement deux gènes responsables de la maladie, le PKD1 et le PKD2, qui codent pour les protéines que sont la polycystine 1 et la polycystine 2, respectivement. Les mutations du gène PKD1 sont impliquées dans la majorité (de 75 à 85 %) des cas de MPRAD8. Ces polycystines sont localisées sur le cil primaire des
cellules de l’épithélium tubulaire rénal et, lorsque leur activité est suffisamment réduite, le signal calcique normal à l’intérieur de la cellule en est perturbé. Il en résulte une prolifération cellulaire, la formation de dilatations kystiques dans le tubule rénal, la sécrétion de fluide et la croissance de kystes, lesquels compriment, puis détruisent irréversiblement le parenchyme rénal normal, d’où le déclin de la fonction rénale. Chez les porteurs de mutations de PKD1, la néphropathie évolue généralement plus rapidement, de sorte que la moitié d’entre eux parviennent au stade de la dialyse au cours de leur sixième décennie de vie, soit 10 ans plus tôt que les porteurs de mutations de PKD2 en moyenne. Il faut cependant préciser que la vitesse à laquelle la maladie évolue varie considérablement d’une famille à une autre et au sein d’une même famille, et que d’autres facteurs de risque cliniques et environnementaux peuvent influencer le cours de la maladie3.
Diagnostic
La méthode classique et principale pour diagnostiquer la MPRAD est l’échographie abdominale. Il existe des critères pouvant être utilisés pour cet examen, sans égard au génotype en cause. Le diagnostic peut être confirmé (valeur prédictive positive de 100 %) chez les personnes âgées de 15 à 39 ans qui sont porteuses d’au moins trois kystes dans un rein ou dans les deux, chez celles de 40 à 59 ans qui présentent au moins deux kystes dans chaque rein et chez celles de 60 ans et plus qui sont porteuses d’au moins quatre kystes dans chaque rein. Lorsque moins de deux kystes rénaux sont décelés chez une personne de plus de 40 ans, le diagnostic de MPRAD peut être exclu avec certitude.
Les conséquences de la MPRAD vont au-delà du déclin de la fonction rénale et de l’évolution vers l’insuffisance rénale.
Cela dit, ces critères s’appliquent uniquement aux personnes ayant des antécédents familiaux de MPRAD10. Dans les cas où les antécédents familiaux sont inconnus ou inexistants, nous ne disposons pas de critères bien validés pour confirmer le diagnostic, mais celui-ci peut être fortement étayé par la présence de nombreux kystes sur les deux reins (c.-à-d. au moins 20) ou par des manifestations d’atteinte extra-rénale. Lorsque le diagnostic est incertain, il peut être bon de soumettre le patient à des tests génétiques, qui peuvent également fournir des renseignements pronostiques sur le risque d’évolution de la maladie2.
Incidence
Les conséquences de la MPRAD vont au-delà du déclin de la fonction rénale et de l’évolution vers l’insuffisance rénale. Il existe d’autres manifestations associées au dysfonctionnement rénal, notamment l’hypertension, la douleur et les malaises rénaux (signalés chez 60 % des patients atteints de la maladie), les calculs rénaux (20 %), les hémorragies kystiques et les infections kystiques. La MPRAD s’accompagne également de manifestations extra-rénales pouvant inclure la maladie polykystique du foie, l’atteinte kystique d’autres organes tels que le pancréas et les voies séminales, l’atteinte valvulaire cardiaque, les hernies de la paroi abdominale et la diverticulose2. Une autre complication importante est la formation d’anévrismes intracrâniens. Cette manifestation, bien que relativement peu courante (fréquence d’environ 8 %), peut avoir des conséquences graves. Le dépistage par imagerie par résonance magnétique (IRM) ou par angiographie par tomodensitométrie est indiqué chez les patients qui présentent des symptômes semblables à ceux d’un AVC ou des céphalées intenses ou atypiques, chez ceux ayant des antécédents familiaux d’anévrisme ou de mort subite, chez les personnes qui exercent une profession risquée (p. ex. pilote d’avion) ou avant une chirurgie lourde ou la mise en route d’une anticoagulothérapie prolongée. Certains pourraient recommander que ce dépistage soit fait systématiquement au moment du diagnostic ou pour rassurer un patient préoccupé par cette éventualité11. Aux effets de la MPRAD sur la santé physique s’ajoutent le fardeau psychologique de se savoir atteint d’une maladie chronique, l’inquiétude pour ses enfants ou face à la possibilité de fonder une famille, la douleur chronique ou l’anxiété soulevée par la crainte de devoir un jour recourir à la dialyse ou à la transplantation rénale. Les effets sur l’assurabilité, les études et le travail peuvent aussi être considérables.
La MPRAD : une maladie évolutive
La MPRAD étant une maladie évolutive, il peut s’écouler plusieurs décennies avant que survienne le déclin de la fonction rénale. Au stade initial de la maladie, la destruction du tissu rénal provoquée par la croissance des kystes est habituellement compensée par l’hyperfiltration glomérulaire. Ce n’est qu’au stade avancé de la maladie (c.-à-d. une fois que le parenchyme rénal a été détruit en grande partie et de manière irréversible) que l’hyperfiltration glomérulaire ne parvient plus à compenser suffisamment et que le DFG commence à chuter rapidement. Pour cette raison, le DFG constitue un marqueur peu sensible de l’évolution de la maladie14. L’hypertrophie des reins ou l’augmentation du volume rénal total peuvent faire l’objet d’un suivi et être utilisées à titre de biomarqueurs précoces d’un éventuel déclin de la fonction rénale et de complications liées à celui-ci7,9. L’évolution naturelle de la MPRAD ne saurait être mieux démontrée que par les résultats de l’étude d’observation prospective que le CRISP (Consortium of Radiologic Imaging Studies of Polycystic Kidney Disease) a menée auprès de 241 patients atteints de MPRAD durant plus de 14 ans, étude qui a été parrainée par les NIH (National Institutes of Health). Au moment de leur admission à cette étude, les patients avaient entre 15 et 45 ans; leur fonction rénale était préservée, mais la plupart d’entre eux présentaient un facteur de risque d’évolution, telle qu’une hypertension ou une protéinurie. L’étude a démontré que les reins ne grossissaient pas à la même vitesse d’un patient à un autre; toutefois, un volume rénal total élevé au départ était prédictif d’un DFG moins élevé après huit ans, et était associé à des complications de la maladie (p. ex. hypertension, hématurie macroscopique et protéinurie)1,6.
Les facteurs qui ont une valeur pronostique et qui semblent avoir un lien défavorable avec une détérioration accélérée des reins comprennent le génotype (maladie liée au gène PKD1 et à une mutation tronquante), des antécédents familiaux d’insuffisance rénale terminale (IRT) d’installation précoce, des signes de déclin prématuré du DFG, l’augmentation du volume rénal total, la présence d’une hypertension prématurée ou de complications urologiques, un taux élevé d’acide urique et une albuminurie ou une protéinurie manifeste13. Par ailleurs, d’après les résultats obtenus lors d’études chez l’animal, une consommation élevée de caféine et une faible consommation d’eau soulèvent des préoccupations.
Options de traitement
Une meilleure compréhension des voies intracellulaires qui stimulent la prolifération et la croissance des kystes a donné lieu à la réalisation de plusieurs études auprès de patients atteints de MPRAD risquant de voir leur maladie évoluer. Les essais cliniques de phase III menés avec répartition aléatoire faisant appel à des inhibiteurs de la cible de la rapamycine chez les mammifères (mTOR), soit le sirolimus et l’évérolimus, n’ont pas fait preuve d’efficacité pour ce qui est de ralentir l’augmentation du volume rénal total et de préserver le DFG.
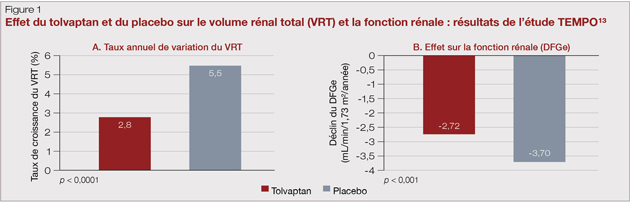
...le traitement actif [avec le tolvaptan] avait ralenti l’augmentation du volume rénal et a laissé suggérer qu’il avait permis de retarder la détérioration du DFG...
Les analogues de la somatostatine sesont révélés prometteurs durant une étude préliminaire. Toutefois, l’étude TEMPO 3:4 (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes), durant laquelle 1 445 patients ont été répartis aléatoirement de façon à recevoir soit du tolvaptan, un antagoniste des récepteurs de la vasopressine-2, soit un placebo pendant trois ans, a révélé que le traitement actif avait ralenti l’augmentation du volume rénal (Figure 1A) et a laissé suggérer qu’il avait permis de retarder la détérioration du DFG (Figure 1B) et d’atténuer la douleur. Les effets secondaires ont été plus nombreux chez les patients ayant reçu le médicament (p. ex. polyurie, soif, goutte et, à souligner, hépatite ayant motivé l’abandon du médicament)5,15 Le tolvaptan a été homologué par Santé Canada et constitue à l’heure actuelle le seul traitement de fond s’offrant aux patients atteints de MPRAD. Par ailleurs, l’essai clinique HALT-PKD (Halt Progression of Polycystic Kidney Disease) ciblait un objectif plus ambitieux pour la tension artérielle (TA mesurée à la maison se situant entre 95/60 mmHg et 110/75 mmHg) chez les patients âgés de moins de 50 ans, atteints de MPRAD, ayant un DFGe de plus de 60 mL/min et ne présentant pas de trouble cardiovasculaire concomitant d’importance notable11.
Il y a lieu d’être très optimistes compte tenu des progrès cliniques déterminants qui ont été réalisés au cours de la dernière décennie, tant au chapitre de la compréhension de la physiopathologie de la MPRAD que de celui des interventions ciblées pouvant modifier le cours de la maladie. Il est donc primordial de dépister la MPRAD chez tous les patients qui en sont atteints, au stade sous-clinique ou peu avancé de la maladie qu’ils soient symptomatiques ou non et chez ceux qui présentent des signes d’hypertrophie rénale avant qu’ils subissent une perte importante de leur fonction rénale. Dans un rapport de consensus d’experts publié au Canada, on recommande que tous les patients atteints de MPRAD soient orientés vers un néphrologue aux fins d’évaluation du risque d’évolution et d’admissibilité au traitement13.
Dans un rapport de consensus d’experts publié au Canada, on recommande que tous les patients atteints de MPRAD soient orientés vers un néphrologue aux fins d’évaluation du risque d’évolution et d’admissibilité au traitement.
Dans bien des cas, les patients exposés à un faible risque d’évolution peuvent être suivis et classés en fonction de leur niveau de risque à intervalles d’un ou deux ans par un néphrologue. Chez ceux qui reçoivent un traitement ciblé ou qui participent à une étude clinique, le suivi en néphrologie peut être plus étroit.
Conclusion
Au cours de la dernière décennie, des pas de géant ont été accomplis dans la compréhension de la physiopathologie et des facteurs de risque d’évolution de la MPRAD. La mise au point de traitements innovants permettant d’agir sur la détérioration rénale donne à ceux qui souffrent de MPRAD une raison d’espérer.
Références :
1. Chapman A, Guay-Woodford L, Grantham J, et coll. Renal structure in early autosomaldominant polycystic kidney disease (ADPKD): the consortium for radiologic imaging studies of polycystic kidney disease (CRISP) cohort. Kidney Int 2003; 64:1035-45.
2. Chapman AB, Devuyst O, Eckardt KU, et coll. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int, 31 juillet 2015; 88(1):17-27.
3. Cornec-Le Gall E, Audrezet MP, Chen JM, et coll. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 2013; 24:1006-13.
4. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med 1993; 329(5):332-42.
5. Gansevoort RT, Arici M, Benzing T, et coll. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA working groups on inherited kidney disorders and European renal best practice. Nephrol Dial Transplant 2016; 31(3):337-348.
6. Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol 2006; 1:148-57.
7. Grantham JJ, Torres VE. The importance of total kidney volume in evaluating progression of polycystic kidney disease. Nat Rev Nephrol 2016; 12(11):667-677.
8. Hateboer N, v Dijk MA, Bogdanova N, et coll. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 study group. Lancet 1999; 353:103-7.
9. Irazabal MV, Rangel LJ, Bergstralh EJ, et coll. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol 2015; 26:160-72.
10. Pei Y, Obaji J, Dupuis A, et coll. Unified criteria for ultrasonographic diagnosis of ADPKD. Clin J Am Soc Nephrol, 1er janvier 2009; 20(1):205-12.
11. Perrone RD, Malek AM, Watnick T. Vascular complications in autosomal dominant polycystic kidney disease. Nat Rev Nephrol, 1er octobre 2015; 11(10):589-98.
12. Schrier RW, Abebe KZ, Perrone RD. Blood pressure in early autosomal dominant polycystic kidney disease. N Eng J Med, 5 mars 2015; 372:975-7.
13. Soroka S, Alam A, Bevilacqua M, et coll. Assessing risk of disease progression and pharmacological management of autosomal dominant polycystic kidney disease: a Canadian expert consensus. Can J Kidney Health Dis, 1er mars 2017; 4:2054358117695784.
14. Tangri N, Hougen I, Alam A, et coll. Total kidney volume as a biomarker of disease progression in autosomal dominant polycystic kidney disease. Can J Kidney Health Dis, 2 mars 2017; 4:2054358117693355.
15. Torres VE, Chapman AB, Devuyst O, et coll. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407-18.